
Placental Development and Early Pregnancy Pathology
Authors
INTRODUCTION
In basic physiologic terms, the placenta is a site of exchange between the maternal and fetal bloodstreams. Nutrients and oxygen are extracted from the maternal circulation, and metabolic byproducts, hormones, and many other molecules are passed into the maternal circulation from the placenta. The placenta transfers many molecules into the fetoplacental pool proportionate to their concentrations in the maternal circulation. The three major modifiers of placental transfer function are maternal blood flow, fetoplacental blood flow, and placental trophoblast membrane permeability. By extension, factors that can limit fetal oxygenation, nutrition, and metabolism are (1) altered maternal perfusion, (2) altered fetoplacental perfusion, (3) reduced placental permeability, and (4) increased placental metabolic needs. Battaglia and Meschia describe the factors determining net fetal nutrient transfer: “… properties of the placenta, such as maternal and fetal blood flows, the pattern of placental perfusion, the surface, thickness and physicochemical properties of the placental membrane, the metabolic activity of the placenta, the various mechanisms of transfer which are available (e.g., diffusion, carrier mediated transfer, active transfer), and regional differences in placental histology and function” (our italics).1 High uterine blood flow, uniform fetal perfusion of the placenta, production of a fetal hemoglobin with greater oxygen affinity than maternal hemoglobin, high fetal cardiac activity, and increased fetal tissue perfusion are the main means by which optimal fetal oxygenation is assured. Normal placental growth and development will maximize placental efficiency while controlling its growth and metabolic needs.
To establish a flourishing intrauterine pregnancy, the trophoblast must anchor to and invade the decidualized endometrium,2 and the uterine vasculature must be able to permit dramatic, progressive increases in blood flow. The implantation process remodels the uterine epithelium and stroma to establish a line of nutrient supply from the maternal tissues and circulation to the conceptus.3 Endometrial invasion has two components, interstitial invasion of the trophoblast into the decidua and stroma, and the endovascular route, by which the vascular remodeling is effected. Interstitial invasion of the endometrium probably involves both an infiltrative and a phagocytic or erosive component.4, 5 The infiltrative process involves a complex orchestration of cell adhesion molecule and integrin sequential expressions. The erosive process likely involves far more primitive molecular signals6 and may be linked to the primitive immune mechanisms represented by the abundance of natural killer cells in the normal late luteal and early pregnant human endometrium.7
The invasive trophoblast is believed to destroy the nonpregnant uteroplacental spiral arterial wall, replacing the media with a nonmuscular, nonelastic (and likely “pressure-passive”) fibrinoid and temporarily serving as the lining cells to the remodeled arterial structure.8, 9, 10 Much uterine vascular remodeling occurs without local trophoblast, however (Fig. 1A). Vascular effects may still be the result of secreted trophoblast products, but some aspects of vascular remodeling may require the immediate presence of trophoblasts. Boyd and Hamilton11 describe marked vascular alterations as the spiral arteries “approach the trophoblastic shell,” including marked endothelial hypertrophy disrupting the normal smooth contour of the vessel lumen, and increasing attenuation of the media to the point that the lining endothelium is surrounded only by a layer of reticular or collagen fibers (Fig. 1B). Some of these early effects may be initiated by maternal (rather than fetoplacental) signals. The better understanding of non-trophoblast-dependent(and potentially pre-implantation) uterine vascular changes may permit improved in vitro fertilization success rates, and fewer later gestational complications.12, 13
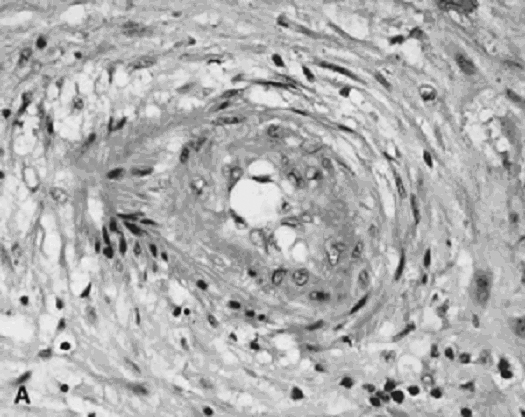 Fig. 1. A. Artery away from the implantation site with myxoid vascular change, 14 weeks’ gestation (hematoxylin-eosin, ×40). B. Normally adapted maternal spiral artery, early second trimester.
Fig. 1. A. Artery away from the implantation site with myxoid vascular change, 14 weeks’ gestation (hematoxylin-eosin, ×40). B. Normally adapted maternal spiral artery, early second trimester.
The trophoblast’s intense vasotropism serves the purpose of establishing, from the earliest days of gestation, a maternal circulation providing the conceptus with nutrients. This circulation may be a sluggish capillary-derived blood pool. The circulation evolves, with spiral arterial remodeling, into a high-volume (capacitance), low-resistance circuit. The repeated failure to identify intervillous circulation both on direct visualization of the intervillous space and using perfused hysterectomy specimens before the 10th to12th week of pregnancy,14, 15 combined with documentation of a low-oxygen environment within the celomic cavity,16 make a standard arterial circulation improbable during embryogenesis. In this schema, too early initiation of maternal arterial perfusion of the intervillous space may damage the conceptus (via oxidative stress)17 and cause early pregnancy loss.18 Others have suggested that fixation artifact and intervillous flow rates (below the current limits of Doppler resolution) may explain most of the flow observations,19 but the evidence for the early embryo’s vulnerability to oxidative injury is also compelling.17 Early pregnancy intervillous flow is not likely a brisk arterial one, and is likely part of a highly unusual period of intensive cellular proliferation and differentiation despite comparative hypoxia.
Retrograde trophoblast extension into spiral vessels commences in the venous circuit, and proceeds against the direction of flow into the arterial tree.20 Trophoblastic remodeling is often considered to proceed in two waves, the first completed by the late first trimester. When the second wave is completed, by the early second trimester,21 invasive trophoblast has penetrated the superficial one third of the myometrium. The “waves” of invasion are probably a continuous process, with a more open-ended completion date. Especially in pathologic pregnancy, endovascular trophoblast can be seen in the basal plate level uteroplacental arteries well into the third trimester. Doppler studies have demonstrated a progressive decrease in the downstream resistance in the uterine circulation from implantation,22 which is paralleled by a fall in umbilical impedance.23 Uterine artery flow increases from 1 to 2% of cardiac output in the nonpregnant state to as much as 30–50% over baseline values by 20–24 weeks’ gestation.24 Complete conversion includes the re-endothelialization of the maternal artery. Failure to complete regrowth of maternal endothelia over the new uteroplacental fibrinoid wall is part of the uteroplacental vascular pathology of preeclampsia.25
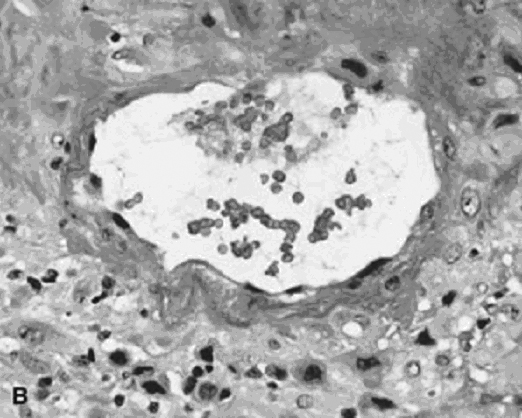
Converted uteroplacental vessels (Fig. 2),25 no longer responsive to vasomotor stimuli, empty into the intervillous space. Nonpregnant spiral arterial flow is a low-volume (capacitance), high-resistance system. After trophoblast remodeling of spiral vessels, the intervillous space is a high-capacitance, low-resistance system (blood pressure of approximately 10–12 mmHg) that is much larger than apparent in the delivered placenta. In a perfused hysterectomy specimen at the eighth month, the intervillous space occupied half the volume of the placenta.26 In the relaxed (noncontracting) uterus, intervillous pressure is similar to intra-amniotic pressure (at approximately 6–10 mmHg).27
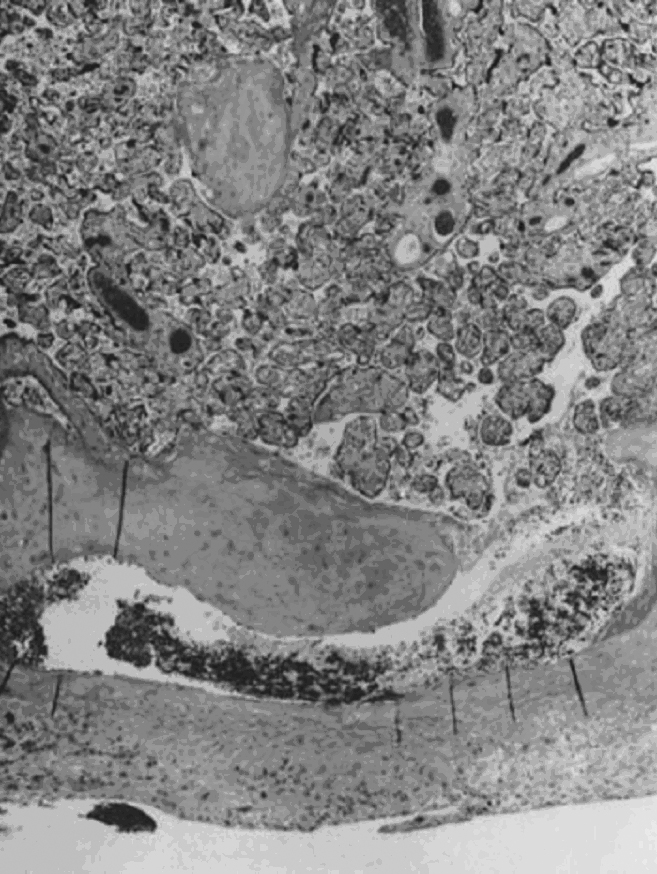 Fig. 2. Base of the placenta, showing a large maternal vessel entering the intervillous space (hematoxylin-eosin, ×4).
Fig. 2. Base of the placenta, showing a large maternal vessel entering the intervillous space (hematoxylin-eosin, ×4).
The uterine arterial vasculature is comparatively and transiently denervated by pregnancy.28, 29 Because the placenta and umbilical cord are not innervated,30 perfusion within the uteroplacental and fetoplacental circulations must depend on anatomic and/or humoral mediators. There is decreased trophoblastic invasion of decidua at the placental margins compared to the center, with less extensive uteroplacental vascular conversion.31 Thus the placental margins are less well perfused, less functional zones that contribute little to placental exchange functions. Routine sampling of placental margins is of limited value.
Three types of trophoblast can be distinguished by their anatomic sites: (1) villous trophoblast, lining the intervillous blood space with cytotrophoblast “stem cells” producing mature syncytiotrophoblast; (2) anchoring (mononuclear) trophoblast that physically anchor villi to the maternal basal plate; and (3) invasive interstitial and endovascular trophoblast that we have already discussed. Interstitial trophoblast cells may fuse (to form multinucleate “placental bed giant cells”) coincidently with the completion of uteroplacental vascular conversion.31, 32 Multinucleate giant cells can be seen in the basal plate of pregnancies spontaneously lost in the late first or early second trimester, before vascular conversion should be complete. In these, we question if early trophoblast fusion is a marker of pathologic maternal-placental interaction (and by extension, uteroplacental vascular conversion). All types of trophoblast differentiate and express antigens in highly coordinated and distinct patterns that are influenced by soluble factors and extracellular matrix components.33, 34 Villous trophoblast does not express class I or class II histocompatibility antigens. Invasive trophoblasts express human leukocyte antigen G (HLA-G), a major histocompatibility antigen, expression of which is restricted to a few cell types, including trophoblast.35 Recent reviews summarize current data regarding regulation and modulation of trophoblast functions in implantation and early placental development.36, 37, 38, 39
Villous morphologic changes in pregnancy are paralleled by placental functional maturation (Fig. 3A). The “primary villi” are columns of cytotrophoblast that extend from the cytotrophoblastic shell. The intervillous space at this age is essentially labyrinthine. Mesodermal invasion into (or in situ differentiation within) primary villi leads to “secondary villi.” The appearance of blood vessels transforms these into “tertiary villi.”40 The mature villous functional unit is a barrel, with the staves formed by the large fetal stem vessels, and the villous tree arborizing toward the center where the most recently formed villi are found.40 The fetal–neonatal postmaturity syndrome is considered to indicate insufficiency of the aging placenta,41 but some placental growth continues until late in gestation.42 If a placenta older than 42 weeks shows healthy terminal villi, and indication of active villous growth, a cause for fetal/neonatal compromise other than “placental insufficiency” may be sought.
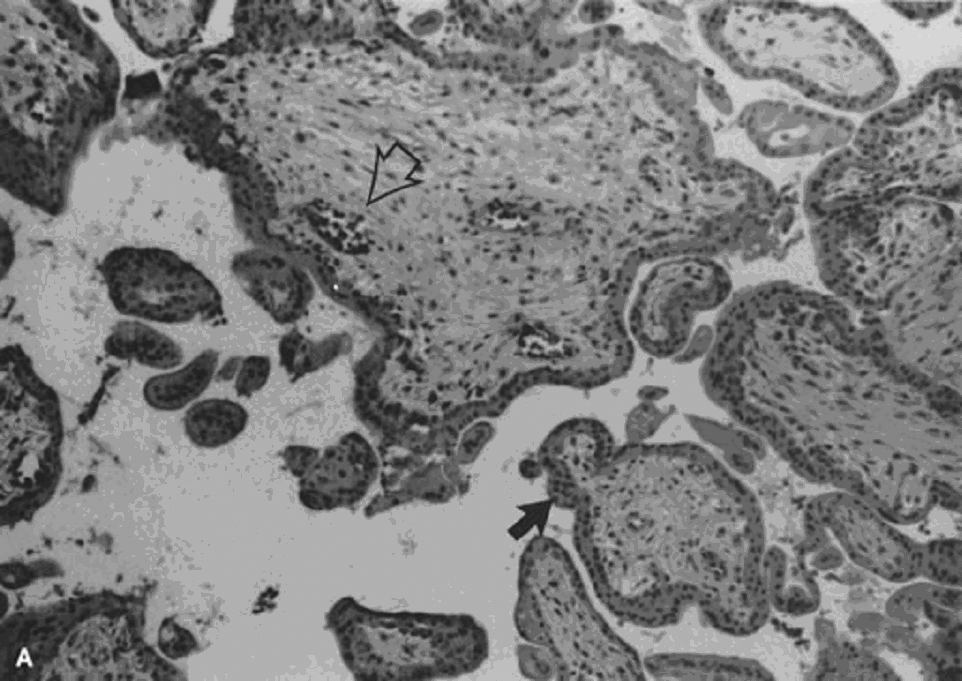 Fig. 3. A. The large villi of the early first trimester have a continuous (black arrow) two-cell layer of epithelium (outer syncytiotrophoblast, inner cytotrophoblast) with loose stroma and sparse, centrally located vessels containing exclusively nucleated erythrocytes (open arrow ) (hematoxylin-eosin, ×10). B. The small terminal villi of a term placenta (hematoxylin-eosin, ×10). C. Vasculosyncytial membrane (arrows) is the hallmark of a term villus, near fusion of the trophoblast basement membrane with the fetal capillary endothelium, with adjacent normal syncytial knots (hematoxylin-eosin, ×40).
Fig. 3. A. The large villi of the early first trimester have a continuous (black arrow) two-cell layer of epithelium (outer syncytiotrophoblast, inner cytotrophoblast) with loose stroma and sparse, centrally located vessels containing exclusively nucleated erythrocytes (open arrow ) (hematoxylin-eosin, ×10). B. The small terminal villi of a term placenta (hematoxylin-eosin, ×10). C. Vasculosyncytial membrane (arrows) is the hallmark of a term villus, near fusion of the trophoblast basement membrane with the fetal capillary endothelium, with adjacent normal syncytial knots (hematoxylin-eosin, ×40).
The beating embryonic heart begins to pump nucleated erythrocytes from the yolk sac throughout the villus circulation by the end of the fifth week of gestation. The proportion of nucleated and anucleate erythrocytes in the fetoplacental circulation is tightly correlated with crown–rump length.43 Villous macrophages (Hofbauer cells) are numerous throughout gestation (40% of stromal cells),44 although at term they may be less conspicuous. Villi arborize by mesenchymal invasion of syncytial sprouts.45 Sprouts can be deported into the maternal circulation, where they may influence maternal tolerance of the conceptus. Villi originally develop over the entire sphere of the conceptus, but atrophy over the extraplacental membranes by the end of the first trimester. Villous atrophy and tissue death related to this normal part of placental development may be difficult to distinguish from pathologic villous necrosis (e.g., infarcts). Remnants of ghost villi can be seen on the free membranes (chorion laeve) even at term. The chorion frondosum develops into the placental disc. Aberrant persistence of proliferating villi distant from the main placenta disk form “accessory” or succenturiate lobes.
The delivered placenta is “deflated” compared to its state in utero, a consideration for anyone attempting ultrasound or anatomic correlations.26, 46 The average diameter of the delivered placenta at term has been reported as 18.5 cm (range 10.5–24.5 cm), with a mean thickness of 2.3 cm (range 1.1–4.1 cm).46 The same source described the early growth of the placenta as primarily an increase in diameter of the chorionic disc, while in the later stages of gestation, placental weight increased primarily due to an increase in placental thickness.46 They likewise reported the mean diameters of the placenta at the third to sixth months as 5.8 cm, 8.2 cm, 10.8 cm, and 13.0 cm, respectively. The number of major villous trunks remains constant;45 therefore, placental growth requires that each major trunk (established in the early months of gestation) must arborize or otherwise develop proportional to the increase in placental weight from early gestation to delivery. Schneider has recently reviewed the ultrasound data regarding placental volume in the midtrimester.47 The mean weekly increase in placental volume between 16 and 24 weeks was 31 ± 8 cm3; further, placental volume in the second trimester was closely correlated with infant birthweight, suggesting that midtrimester placental well-being is an important determiner of late fetal growth.
Early villous vessels are situated deep in the villous stroma, at a mean distance of 24–27 μm from the intervillous space to villous vessel lumen (Fig. 3A).45 Placental mass increases more slowly than fetal weight by the end of the second trimester (Table 1).48 In the third trimester, fetal growth is able to continue disproportionate to the mass of the nurturing placenta because the later placenta has increased diffusion efficiency. Progressive reduction in trophoblast thickness, villous size, and the distance between the intervillous space and the fetoplacental capillary are the anatomic bases for improved transfer efficiency (Fig. 3B). The placental vasculature also increases in complexity, with increasing numbers of capillary outlines per villus, until about 36 weeks’ gestation. Additionally, there is a massive enlargement of the syncytiotrophoblastic surface, disproportionate to syncytial volume. Microvillous surface increases until about 36 weeks’ gestation. Overall, this increases the villous surface area of exchange.49, 50, 51
Table 1. Expected fetal and placental weight ratio
| Weeks' gestation | Placental wt (g) | Fetal wt (g) | F:P ratio |
| 8 | 6 | 1.1 | 0.18 |
| 10 | 14 | 5 | 0.36 |
| 12 | 26 | 17 | 0.65 |
| 14 | 42 | 30 | 0.71 |
| 16 | 65 | 60 | 0.92 |
| 18 | 90 | 130 | 1.44 |
| 20 | 115 | 250 | 2.17 |
| 22 | 150 | 400 | 2.67 |
| 24 | 185 | 560 | 3.03 |
| 26 | 210 | 750 | 3.57 |
| 28 | 250 | 1000 | 4.00 |
| 30 | 285 | 1260 | 4.42 |
| 32 | 315 | 1550 | 4.92 |
| 34 | 355 | 1900 | 5.35 |
| 36 | 390 | 2300 | 5.90 |
| 38 | 425 | 2750 | 6.47 |
| 40 | 470 | 3400 | 7.23 |
(From Molteni RA: Placental growth and fetal/placental [F/P] ratios throughout gestation: their relationship to patterns of fetal growth. Semin Perinatol 8(2):94, 1984)
Placental permeability, the ease of transfer across the placental membrane, increases even after placental growth rate peaks by 30–32 weeks’ gestation.1 This is believed to be due to the formation of “vasculosyncytial membranes.” In a vasculosyncytial membrane, villous capillaries abut the trophoblast basement membrane, and the trophoblast nuclei and cytoplasm are clumped at the edge of the capillary margin (Fig. 3C). The membrane itself measures only 0.5–1.0 μm in thickness.45 Mean diffusion distance between the mother’s bloodstream and the fetal circulation is ~3 μm.50 Progressive improvement in placental permeability leads to a “flow-limited” placental transfer capacity at term.1
In vivo studies in humans have identified a decrease in the volume of umbilical venous flow in the third trimester.52 Therefore, despite the remodeling of the placenta for increased functional capacity, there is a real potential for decreased fetal supply during the last trimester. At term the weight-specific placental oxygen consumption is almost five times as high as that of the fetus.53 Shortly before birth, the fetus also begins to prepare for extrauterine life by switching synthesis from hemoglobin F (HbF) to hemoglobin A (HbA). Although HbF has a higher oxygen affinity than HbA, which confers an “advantage” to the fetus favoring fetal oxygen extraction from maternal intervillous blood, late in pregnancy this advantage is reduced. These features combine to create a physiologic basis for “placental insufficiency” late in pregnancy and post term as fetal and placental growth, fetal and placental metabolic needs, and the maturational changes in Hb combine potentially at the upper limits of uterine supply.
Placental physiology is not the primary focus of this chapter. Brief highlights of specific aspects of placental physiology are presented here concerning maternal fetal placenta water balance and placenta respiratory function. The placenta is the major determiner of fetal water balance,54 with a relatively insignificant contribution of the fetal kidney. Maternal dehydration is directly paralleled by increased fetal plasma osmolality.55 However, the composition of fetal urine is significantly different from that of fetal plasma, indicating a fetal renal role in ion homeostasis. In the absence of fetal stress, fetal urine (and amniotic fluid) is generally hypotonic.56 Amniotic fluid composition is changed from that of fetal urine as it leaves the kidney via gestational age-dependent changes in extraplacental membrane permeability.56
Oxygen and carbon dioxide are exchanged across the placenta by simple diffusion. Longo has summarized the major factors affecting oxygen transfer.57 The placenta is a very metabolically active tissue, consuming one third to one half of the oxygen presented to the intrauterine tissues.1 Oxygen exchange almost always is flow limited, rather than diffusion limited.57 The fetus may direct placentally supplied nutrients, including oxygen, to different tissue uses at different gestational ages.1 Until 70 torr, maternal arterial partial pressure of oxygen (PO2) has little effect on placental oxygen exchanged; below this value, progressively less oxygen crosses to the fetus. Umbilical venous oxygen tension is generally 10–15 torr less than uterine venous blood values. Fetal carbon dioxide production is about equal to PO2 utilization. Fetal carbon dioxide rapidly diffuses into maternal blood depending on the concentration gradient between fetal and maternal blood. Fetal partial pressure of carbon dioxide (PCO2) drops with maternal hyperventilation and rises with maternal hypoventilation, impaired placental perfusion, or permeability.1 Fetal arterial PCO2 can rise during fetal underperfusion of the placenta (or cord compression).1
Carrier-mediated transfer of many molecules across the placenta (including glucose and amino acids) is affected by changes in either maternal or fetal perfusion. The placenta metabolizes most of the glucose obtained from the maternal circulation, and provides the fetus with less moles of glucose than required to fuel the fetus.1 Placental lactate is a major fuel of fetal metabolism. In fetal growth restriction (FGR), fetal nutrient utilization may decrease,58 leading to underutilization of placental lactate and a lactic acidosis. Under conditions of asphyxia, the fetus itself is a net producer of lactate.1 Ketoacids, such as produced in maternal diabetic ketoacidosis, may reduce fetal brain metabolism.1
PLACENTAL EXAMINATION, SELECTION, AND METHODOLOGY
All placentas should be handled according to standard biohazard precautions. Gross examination can be performed on fresh specimens, or after 1–2 days of fixation in formalin. Prolonged refrigeration (5–7 days) does not affect histology. We prefer gross examination of fresh placentas, and select tissue samples that are subsequently fixed in formalin overnight before trimming or processing. Placentas from stillborn fetuses should not be fixed in formalin on receipt because placental fibroblasts may be needed for cytogenetic or other metabolic analyses. Microbial cultures should be performed in the labor and delivery suite, and not hours after birth in the pathology laboratory.
Placentas must be weighed and measured. Different approaches to placental weighing (removing cord, removing extraplacental membranes, draining maternal intervillous blood) may be taken, but one method must be used consistently, and documented in the pathology report. The cord length and diameter are recorded. The fetal–placental weight ratio increases throughout gestation, and abnormally high or low ratios are likely indicators of fetoplacental pathology (see Table 1).
A sample gross dictation form and suggested criteria for selection of placentas for pathologic examination are presented in Tables 2 and 3.
Table 2. Suggested standard dictation for singleton placenta
- Received fresh/fixed/not specifically labeled placenta is a _g placenta with a _g attached membranes and umbilical cord.
- The placenta is fragmented/round/oval/bilobate/accessory lobes and measures _cm × _cm × _cm in thickness.
- Membranes are complete/incomplete and site of rupture is indeterminate/at the placental margin/less than 10 cm/greater than 10 cm, from the nearest placental margin.
- Membranes are translucent/thickened/shiny/granular/green/yellow/foul smelling, with squamous metaplasia/amnion nodosum/hemorrhage.
- Marginal decidual necrosis is unremarkable/prominent/excessive.
- Intramembranous blood vessels are absent /present /intact ruptured and traverse unsupported for _ cm in the membranes.
- Membranes are inserted marginally/circummarginate/circumvallate over _% of the placental circumference.
- Maximal extent of placenta extrachorales is _cm.
- The umbilical cord has central/eccentric/markedly eccentric/marginal /velamentous insertion, _cm from the nearest placental margin and measures _cm in length and _cm in diameter at the fetal end and _cm in diameter at the placental end.
- The cord has true knot /hematoma/stricture /torsion/thrombosis not associated with hemostat marks, located_.
- The cut surface of the cord shows _ vessels with normal/edematous/decreased Wharton's jelly and no/venous/arterial thrombi.
- The chorionic plate vessels are tortuous/ectatic/thrombosed/calcified and subchorionic fibrin deposition is normal/increased/decreased, with the largest plaque measuring _cm × _cm × _cm in thickness.
- There is/is not grossly obvious missing placental tissue.
- There is fibrinoid deposition decidual/necrosis/calcification/blood clot covering approximately _ % of the maternal surface.
- The cut surface is pale/congested/firm/gritty/edematous and shows fibrinoid/infarction/thrombi occupying approximately % of the substance.
- Miscellaneous findings.
- The standard placental sections are submitted.
- Additional sections of grossly identified abnormalities are submitted as follows.
- Individual lesions are measured.
Table 3. Which placentas should be examined by a pathologist?
Recommended Maternal Indications for Placental Examination
Systemic disorders with clinical concerns for mother or infant (e.g., severe diabetes, impaired glucose metabolism, hypertensive disorders, collagen disease, seizures, severe anemia (<9 g)
Premature birth at 34 weeks' gestation or earlier
Intrapartum fever and/or infection
Unexplained third trimester bleeding or excessive bleeding >500 mL
History of HIV, syphilis, cytomegalovirus, primary herpes, toxoplasmosis, or rubella infections during this pregnancy
Severe oligohydramnios
History of unexplained or recurrent pregnancy complication fetal growth restriction, stillbirth, spontaneous abortion, premature birth)
Poor pregnancy outcome associated with intrauterine intervention, either diagnostic or therapeutic, for fetal abnormality or induced abortion for fetal abnormality
Abruption
Abnormal antenatal testing leading to therapeutic intervention
Optional Maternal Indications for Placental Examination
Premature birth at 35–36 weeks' gestation
Severe unexplained polyhydramnios
History of substance abuse
Recommended Fetal/Natal Indications for Placental Examination
Admission to intensive care nursery or transfer to another facility for care
Stillbirth/Perinatal death
Compromised clinical condition defined as any of the following:
Cord blood pH <7.0
Apgar score <7 at 5 minutes
Ventilatory assistance >10 minutes
Severe anemia (hematocrit <35%)
Hydrops fetalis
Birthweight <10th centile
Seizures
Infection/sepsis
Major congenital anomalies, dysmorphic phenotype, or abnormal karyotype
iscordant twin growth or vanishing twin
Multiple gestation with like sex infants and fused placentas
Optional Fetal/Natal Indications for Placental Examination
Birth weight >95th centile
Discordant growth parameters in any infant
Multiple gestation without other indication
Recommended Placental Indications for Placental Examination
Visible or palpable abnormality (e.g., infarct, mass, vascular thrombosis, retroplacental hematoma, amnion nodosum, abnormal coloration or opacification)
Small-for-gestational-age placental dimensions or weight
Umbilical cord lesions (e.g., thrombosis, torsion, true knot, single artery, absence of Wharton's jelly)
Viscid/thick meconium
Short umbilical cord (<32 cm at term)
Optional Placental Indications for Placental Examination
Large-for-gestational-age placenta
Abnormalities of placental shape
Long umbilical cord (>100 cm)
Marginal or velamentous cord insertion
(Adapted from Langston C, Kaplan C, Macpherson T et al: Practice guideline for examination of the placenta: Developed by the Placental pathology practice Guideline Development Task Force of the College of American Pathologists. Arch Pathol Lab Med 121 (5):449, 1997)
The placental parenchyma is grossly examined by slicing the maternal surface perpendicular to the chorionic plate at approximately 1-cm intervals, leaving the chorionic plate intact. Each surface of each slice is examined for obvious lesions (e.g., infarct, intervillous thrombi) or subtle lesions, such as increased villous granularity (e.g., villous swelling or chronic villitis). Villous tissue should be uniform in color; dark or pale areas may mark microscopic pathology.
We suggest the following approach to placental sampling:
- One cassette containing one section from the fetal end of the umbilical cord (the end closest to the baby), 2.5 cm from the insertion (to ensure accurate vessel count) and avoiding areas with evidence of cord clamping, and one strip of extraplacental membranes taken from the rim of the site of rupture (if identifiable).
- One cassette containing one section from the placental end of the umbilical cord (the end closest to the chorionic plate), and one strip of extraplacental membranes extending from the rim of the site of rupture perpendicular to the placental margin. If a few marginal villi are included in this strip, this piece of membrane will always be able to be distinguished from the site of rupture sample. (Cassettes 1 and 2 may be combined.)
- Four sections containing grossly representative villous parenchyma in the central 60% of the chorionic disc (avoiding the margins) and can include areas of pallor and increased granularity. Full-thickness sections (i.e., extending from the chorionic plate to the basal plate) are preferable, but often the placenta is too thick. In such cases, the full-thickness section can often be divided, and the pieces placed in a single cassette. At minimum, these four tissue samples should include:
One cassette containing chorionic plate from an area with minimal subchorionic fibrin, and containing chorionic vessels.
Three cassettes based on the basal plate. Examination of the uteroplacental vascular segments delivered with the placenta can be readily achieved, especially in the fixed placenta, in which they can often be seen as small irregularities or an S-shape on the maternal surface. Several thin slices of the basal plate may be placed in one cassette, and will generally yield at least one uteroplacental vessel for examination. These cassettes also provide chorionic villi from the central areas of the villous parenchyma, representative of the best functioning villous tissue. Additional sections from lesions.
Four sections of placental parenchyma will yield 95% of villous abnormalities. Adequate placental diagnosis can be done with three to four cassettes, depending on the thickness of the placenta. Uteroplacental vessels should be available for review in all cases, especially when uteroplacental vascular pathology is suspected.
PATHOLOGY OF EARLY PREGNANCY LOSS
In the simplest terms, normal fetal growth and development requires a normal fetal developmental program (i.e., a genetically normal conceptus). Abnormal karyotype—the presence of a developmental program manual with extra, missing, or rearranged pages—commonly precludes embryo/fetal development. If the fetus is viable, pregnancy success depends on the physiologic adaptation of the maternal spiral arteries (to allow dramatic increases in volume of uteroplacental blood flow at a pressure that is not traumatic to tissues), and on the development of a functional fetoplacental circulation. Evaluation of the products of conception should include a confirmation of intrauterine pregnancy, but should also assess why the pregnancy failed (see below).
Molar Gestations
Approximately 50% of spontaneous abortions are caused by fetal karyotypic abnormalities. Molar gestations are extreme cases of aneuploidy, which are commonly lethal for the fetus, and diagnosable by routine light microscopic examination. The World Health Organization—International Society of Gynecologic Pathologists has provided guidelines for the classification of complete hydatidiform mole (CHM), partial hydatidiform mole (PHM), and invasive mole.59
Hydatidiform mole (HM) is a nonspecific term that includes two entities: complete HM and partial HM, typical features of which are listed in Table 4.60, 61, 62, 63, 64 The diagnosis of HM should be used only if a distinction between complete and partial HM cannot be made by gross, histologic, and karyotypic data. Clinically, all forms of HM are characterized by elevations of serum human chorionic gonadotropin levels. HM occurs in the United States in 1/2000 deliveries, but worldwide, the incidence varies from 1/200 (Mexico) to 1/4369 (Paraguay), and is more common in women of Asian descent.45 Partial HM is underdiagnosed and is probably more common than complete HM. A differential diagnosis includes hydropic abortus in chromosomally normal or abnormal conceptuses. Risk for choriocarcinoma or recurrence of HM in a subsequent pregnancy differs depending upon the type of molar pregnancy. Adequate sectioning, a minimum of 5 to 10 blocks of placental tissue and anything suspicious of membranes or fetal tissues, is recommended.65, 66, 67 Suction curettage and formalin fixation collapses hydropic villi. A brief submergence in water will rehydrate them and clarify the gross. Many new techniques have been used to differentiate these disorders, but the hallmark is still light microscopy, correlated with gross and clinical features, immunohistochemistry for placental proteins and cell proliferation markers, and determination of parental chromosome by polymerase chain reaction analysis.68
Table 4. Molar gestations: clinical, gross, microscopic, and flow cytometric features
Complete hydatidiform mole | Partial hydatidiform | Hydropic | |
Maternal age | <20, >40 | 27 | n/r |
Weeks gestation | 12 | 15 | 11 |
Clinical diagnosis | Mole | Missed abortion, <10% mole | Sporadic abortion |
Hyperemesis | 50% | Rare | No |
Theca luteum cysts | 20–50% | Rare | No |
Preeclampsia | 25% | Rare | No |
Ultrasound | Snow storm | Rare cysts | Rare cysts |
BhCG mIU/mL | 160,000 | 27,000–80,000 | 19,000 |
Gross | |||
Villi | Yes | Yes | Yes |
Gross vesicles | All | Focal | Rare |
Vesicle size | 5–20 mm | 3–6 mm | 3mm |
Fetus | No | Yes | Yes |
Amount of tissue | 200 g | 150 g | 50 g |
Membranes | No | Yes | Yes |
Microscopic | |||
Villous size | Uniform | Dual population | Uniform |
Villous shape | Round | Scalloped | Round |
Cisterns | Many | Few | Rare |
Stroma | Necrobiosis | No necrobiosis, trophoblastic inclusions | Necrobiosis |
Vessels | Rare | Yes | Yes |
NRBC | No | Yes (>50%) | Yes |
Trophoblast proliferation | Moderate–marked | Minimal–moderate | Atrophic/polar |
Location | Circumferential | Focal | Polar |
Necrosis | Yes | Yes | No |
Lacy vacuolation | Yes | Yes | No |
Karyotype | 96% 46, XX, 4% 46,XY | 58–70% 69, XXY, 27–40% 69, XXX, 2–4% 69, XYY | 20% abnormal |
Flow cytometry | 60–80% diploid, 2–43% tetraploid, 10% aneuploid, rare triploid | 80–100% triploid, 0–20% diploid, 10% aneuploid, 2% tetraploid, 2% haploid | 80–85% diploid, 6–10% triploid, 4% aneuploid, 4% tetraploid |
Risk for malignancy | |||
Recurrence | 1% | Possible | Unknown |
Persistent disease | 10–20% | 4–13% | Unknown |
Choriocarcinoma | 2–8% | Case reports | 1/160,000 |
(From Popek EJ: Partial hydatidiform mole. ASCP Check Sample AP 94–7 (AP-241). American Society of Clinical Pathologists, 1994)
Complete hydatidiform mole develops when an empty ovum is fertilized by two sperm (heterozygotic, dispermic diandrogenetic fertilization) or by one diploid sperm (homozygotic, monospermic, diandrogenetic fertilization).62 Complete HM is classically described as a mass of grapelike vesicles. Histologically, enlarged villi show central edema and circumferential proliferation of syncytiotrophoblasts and intermediate and cytotrophoblasts. Nuclear atypia may be considerable but is without prognostic significance.69 A fetus is generally absent unless there is a concurrent nonmolar twin pregnancy. Residual blood vessels empty of erythrocytes are common in HM villi before 10 weeks' gestation. Early complete HM may be difficult to diagnose.70 Risk of persistent disease and transformation into choriocarcinoma is 10% to 20% and 2–8%, respectively. There may be increased risk of choriocarcinoma in the 6% to 9% of complete HM that result from, dispermic, fertilization.67, 71, 72, 73
Partial hydatidiform mole, incorrectly referred to as a “transitional mole,” is the result of fertilization of a normal ovum by either two sperm (dispermic, diandrogenetic fertilization) or one diploid sperm (diandrogenetic fertilization).62 The tissue may be more abundant than expected for gestational age and usually includes membranes, rarely an umbilical cord or fetus. Only some villi are enlarged, resulting in a dual villous population (Fig. 4A and B). Variable syncytiotrophoblastic proliferation is the key to diagnosis. The diploid partial HM is reported to be more aggressive, yet some still question exactly what it may represent. Some clearly are complete HM, from early curettages in which the villous morphology has not completely developed, and others are twin gestations with a normal fetus and coexistent molar gestation.74, 75 A diploid ovum may be fertilized by a normal haploid sperm (digynic fertilization), resulting in a triploid abortus without partial molar features (Fig. 4C).73 Choriocarcinoma is a rare complication of partial HM. The several case reports76, 77, 78 often fail to completely describe the preceding molar lesion.
|

Hydropic abortus must be included in the differential diagnosis of molar gestation. The classic features are amphophilia of the villous stroma and atrophy of the trophoblasts. The fetoplacental circulation is generally absent or collapsed. Trophoblastic proliferation, if present, is polar, representing a normally implanting villus.
Early Nonmolar Pregnancy Loss
Nonmolar early pregnancy losses include nonmolar aneuploid conceptions and euploid fetuses (potentially viable, although single gene defects will be not identified by crude karyotype). The following working guides help frame our histologic analysis of products of conception and fetal loss.
Findings need to be interpreted in the context of that patient's past reproductive history. To evaluate a sporadic abortion, the most important clinical distinction may be whether the pregnancy was likely euploid (and potentially viable) or not. Euploid pregnancy loss carries a maternal age-independent potential for recurrence, whereas recurrence of aneuploid loss tends to be tied to maternal age.
Ploidy is less important if the patient has had three or more losses and is under 40 years of age, when the risk of serial aneuploid conceptions would be quite low. A recurrent aborter may have a sporadic aneuploid loss, but the critical point is whether any consistent pattern of injury persists across pregnancy losses and independent of ploidy.
Pregnancy loss will be caused either by aneuploidy of the conceptus (pathology intrinsic to the conceptus), or by pathology extrinsic to the embryo/fetus (identifiable in the decidua and/or placenta). This also differentiates pregnancy loss by recurrence risk. Pregnancy failure may be caused either by a nonviable embryo/fetus, or by a decidual/placental environment incapable of supporting conceptus development. If the embryo/fetus is nonviable, then histopathology will reflect early embryo failure, with karyorrhectic (or absent) fetoplacental circulation. If the embryo/fetus is genetically viable, the cause of pregnancy failure may be observed or inferred from patterns of placental and/or decidual injury.
“End-stage” pathology must be carefully distinguished from that potentially causing fetal death. In pregnancy loss, microscopic changes may begin to accumulate without clinical symptoms. By the time a pregnancy loss becomes clinically symptomatic (i.e., bleeding, cramping), an end-stage pathology (reflecting tissue degeneration common to all etiologic types of pregnancy loss) would be expected to close off blood vessels and allow tissue detachment from the uterus. Coagulation of maternal blood flow and altered inflammatory patterns are part of this final-common-pathway histology of miscarriage.
Fetal viability may be estimated by the type of erythroid elements in the placental circulation. Nucleated erythrocytes are produced in the yolk sac and circulate in the early weeks of pregnancy. With hepatic hematopoiesis (weeks 7–9), anucleate erythrocytes begin to circulate. The proportion of nucleated erythrocytes is closely correlated with crown–rump length (Table 5).43 If the gestational age at fetal demise is known, the observed proportion of nucleated erythrocytes can be compared to the proportion expected for gestational age. A greater proportion of nucleated erythrocytes than expected for fetal age indicates abnormal hematopoiesis, such as seen in chromosomal aberrations (in particular triploidy and trisomies), or hematopoietic stress (e.g., hydrops fetalis, antiphospholipid antibody associated massive placental infarction, and recurrent midtrimester losses with dense chronic intervillous inflammation).
Table 5. Correlation of crown–rump length (CRL) and calculated gestational age (EGA) with estimated percentage nucleated red blood cells (NRBC)
CRL (mm) | EGA (days) | Estimated % NRBC |
10 | 48 | 97 |
15 | 53 | 75 |
20 | 59 | 58 |
25 | 63 | 42 |
30 | 69 | 28 |
35 | 71 | 18 |
40 | 76 | 10 |
45 | 78 | 4 |
50 | 81 | 1 |
(From Salafia CM, Weigl CA, Foye GJ: Correlation of placental erythrocyte morphology and gestational age. Pediatr Pathol 8:495, 1988)
Many histologic features can be evaluated in the placenta and decidua of early pregnancy loss. Those features that could be reliably distinguished in a routine clinical diagnostic service of nonrecurrent (sporadic) spontaneous loss are listed in Table 6.79 Patients had either their first or second loss karyotyped, and therefore did not meet criteria for “recurrent abortion.” Molar gestations were excluded and karyotype was grouped as euploid and aneuploid. The better preserved the fetal circulation, the greater the apparent age at fetal demise, the greater the likelihood of a euploid conceptus. The presence of villous infarct, chronic intervillositis, and chronic uteroplacental vasculitis were each independently associated with a euploid conceptus. From this we suggest that age at death can routinely be assessed by proportion of nucleated erythrocytes. The greater the fetal age assessed by the circulation, the greater the likelihood of euploid conceptus.
Table 6. Villous (N = 221) and decidual (N = 175) features of nonrecurrent spontaneous losses with normal (euploid) and abnormal (aneuploid) chromosome number
Villous | Decidual | Euploid | Aneuploid |
Villous circulation | Absent circulation | 43 (46%) | 7 (59%) |
Moderate/severe degeneration | 24 (26%) | 38 (29%) | |
Mild degeneration | 14 (16%) | 15 (11%) | |
Intact circulation | 9 (10%) | 1 (0.07%) | |
% Nucleated erythrocytes* | 100% (<7 wk)* | 48 (53%)* | 105 (80%)* |
>50% (7–9 wk)* | 7 (8%)* | 8 (6%)* | |
5–50% (9–11 wk)* | 5 (5%)* | 9 (7%)* | |
<5% (>11 wk)* | 30 (33%) | 9 (7%)* | |
Hydropic changes | Not present | 41 (46%) | 42 (32%) |
Mild/moderate | 31 (35%) | 56 (43%) | |
Severe | 18 (20%) | 33 (24%) | |
Perivillous fibrin deposition | Mild | 33 (37%) | 50 (39%) |
| Moderate | 50 (55%) | 72 (55%) |
Severe | 7 (8%) | 9 (7%) | |
Villous infarct* | Not present* | 76 (84%)* | 125 (95%)* |
Present* | 14 (16%)* | 6 (6%)* | |
Chronic intervillositis* | Not present* | 77 (86%)* | 122 (93%)* |
| Present* | 13 (14%)* | 9 (7%)* |
Uteroplacental vascular adaptation | Only normal vessels | 36 (48%) | 56 (56%) |
| Any abnormal | 31 (18%) | 33 (18%) |
Uteroplacental thrombosis | Not present | 47 (63%) | 72 (72%) |
| Present | 28 (37%) | 28 (28%) |
Uteroplacental vasculitis* | Not present* | 43 (57%)* | 68 (68%)* |
| Present* | 32 (43%)* | 32 (32%)* |
Excessive decidual necrosis | Not present | 42 (56%) | 58 (58%) |
| Present | 33 (44%) | 42 (42%) |
Decidual plasma cells | Not present* | 72 (96%)* | 96 (96%)* |
| Present * | 3 (4%)* | 4 (4%)* |
Decreased invasive trophoblasts | Not present* | 43 (57%) | 56 (56%)* |
| Present* | 52 (43%)* | 44 (44%)* |
Significant differences (p < 0.05) between pregnancy losses with normal and abnormal chromosome numbers.
(Salafia CM, Maier D, Vogel C et al: Placental and decidual histology in spontaneous abortions: detailed description and correlations with chromosome number. Obstet Gynecol 82:295, 1993)
Fetal gestational Significant differences (p < 0.05) between pregnancy losses with normal and abnormal chromosome numbers. (From Salafia CM, Maier D, Vogel C et al: Placental and decidual histology in spontaneous abortions: Detailed description and correlations with chromosome number. Obstet Gynecol 82:295, 1993)
Fetal gestational age at death can routinely be assessed by proportion of nucleated erythrocytes. The greater the fetal age assessed by the circulation, the greater the likelihood of euploid conceptus.
Specific decidual and placental lesions that implicate the maternal vasculature and/or chronic intrauterine inflammation are associated with euploid pregnancy loss.
Those decidual and placental features not associated with ploidy represent the end-stage, common pathology of impending abortion.
In our hands, no single feature is as useful as the equation that included all histologic features, maternal age, and the gestational age at loss calculated from the last menstrual period. This summary equation could estimate an 88% likelihood of aneuploidy, and a 96% likelihood of euploidy. Some have not confirmed the utility of specific lesions to diagnose aneuploidy.80, 81 Others find that specific features are associated with aneuploidy but with poor specificity.82, 83 A recent study of 668 characterized abortions obtained positive predictive values of >85% for euploidy for “autoimmune markers, chronic intervillositis, and increased perivillous fibrin with intermediate trophoblast.”83 Together with our study of sporadic, nonrecurrent loss, these studies of recurrent loss confirm the utility of placental diagnoses in ploidy estimation in early pregnancy loss.
Why is estimation of likely ploidy clinically useful? Shifting the balance of likelihood of aneuploidy and euploidy from the statistical 50/50 true in the first trimester to “more likely” or “less likely” improves assessment of future pregnancy risks. There is increased risk of chromosomally normal pregnancy loss after an abortion with a normal karyotype.84, 85 Cheap determination of likely loss of euploid conceptions may permit earlier evaluations, without waiting for the third miscarriage. The same consistency in karyotype of loss can be seen among certain couples with recurrent aneuploid pregnancy loss.84, 86 Such patients may, at minimum, merit a second look at maternal triple screen results in their next pregnancy or consideration of amniocentesis. Nondisjunction has the same statistical probability of occurring in a recurrent aborter with antiphospholipid antibodies as in another woman the same age without recurrent loss. An aneuploid pregnancy loss does not, therefore, exclude the diagnosis of a chronic condition that would cause loss of euploid pregnancies as well.
The linking of uteroplacental vascular and/or chronic inflammatory pathology to euploid pregnancy loss are consistent with associations of placental infarcts and, uteroplacental vasculopathy to maternal autoimmunity,87 and chronic placental inflammation to recurrent fetal loss.88, 89 Subclinical maternal autoimmunity has been identified in at least a subset of patients with otherwise unexplained abortion.90, 91 A major site of the pathology of the antiphospholipid antibody syndrome is the (maternal) uteroplacental vasculature.92 However, both histology and in vitro work suggest that the placenta is also a target of maternal immune-associated damage.92, 93 In women receiving therapy for recurrent spontaneous abortions, fetal karyotype combined with placental histopathologic assessment is helpful in deciding if treatment has failed or if that particular pregnancy would have been lost despite treatment. Pathologic examination of products of conception (POC) should include but not be limited to tissue documentation of the fact of pregnancy. Because the placenta may remain viable long after fetal death (as in cases of “blighted ovum”), villous trophoblast and stroma may be viable for weeks (and capable of direct cytogenetic analysis even if the embryo–fetus is dead).
Confined placental mosaicism is a genetic circumstance not neatly classified by the above schema. An early divergence of cell lineage between placenta and fetus may lead to discordance of the genetic composition of the trophoblast and the conceptus;94 placental post-mitotic errors may also lead to confined placental mosaicism. Kalousek has described three different forms of confined placental mosaicism.94 Direct (trophoblast) analysis of chorionic villous samples may not reflect fetal karyotype. The subject of mosaicism is outside of the scope of this review.
Other more subtle features of placental dysmorphogenesis have been described and ascribed to specific conceptus karyotypes.95 These criteria have been difficult to apply in a clinical laboratory setting, but provide evidence that placental development is affected by karyotypic abnormalities.
Aneuploidy is a less common but still significant finding in pregnancy loss after the first trimester. Liveborn infants are often dysmorphically anomalous; their maldevelopment is paralleled by relatively poor intrauterine growth symmetrically involving both the fetus and placenta. Most profound intrauterine growth restriction is seen in trisomy 18, with trisomy 13 and trisomy 21 showing progressively less severe effects. Most autosomal aneuploidies or heteroploidies are associated with growth-restricted infants.
Trisomic placentas may have irregularly shaped villi and atypical stromal migrating trophoblasts.96 There are fewer small muscular arteries in the stem villi and reduced small muscular artery/villous ratio, but absolute villous count is normal.96 The villi are immature, with histology lagging about 4 weeks. A single umbilical artery is frequent in trisomy 18. In monosomy X, villi tend to be small (with resultant apparent increased intervillous space) and either acellular and fibrotic or irregularly hypercellular. The villous trophoblastic layer is hypoplastic with infrequent syncytial budding. Triploid conceptions, especially those with a diploid maternal genome (digyny), may be nonmolar. In these cases there is usually scant tissue (less than expected for dates). Microscopic features may be relatively normal without trophoblastic proliferation. Tetraploid vili are large, round, and without trophoblastic proliferation.
REFERENCES
Battaglia FC, Meschia G (eds): An Introduction to Fetal Physiology. London, Academic Press, 1986 |
|
Feinberg RF, Kliman HJ, Lockwood CJ: Is oncofetal fibronectin a trophoblast glue for human implantation? Am J Pathol 138: 537, 1991 |
|
Lessey BA: The role of the endometrium during embryo implantation. Hum Reprod 15 (Suppl 6): 5, 2000 |
|
Choy MY, Manyonda IT: The phagocytic activity of human first trimester extravillous trophoblast. Hum Reprod 13 (10): 2941, 1998 |
|
Manyonda IT, Choy MY: Collagen phagocytosis by human extravillous trophoblast: potential role in trophoblastic invasion. J Soc Gynecol Investig 6 (3): 158, 1999 |
|
Greenberg S: Diversity in phagocytic signaling. J Cell Sci 114: 1039, 2000 |
|
Loke YW, King A: Immunology of implantation. Baillieres Best Pract Res Clin Obstet Gynaecol 14 (5): 827, 2000 |
|
Pijnenborg R, Dixon G, Robertson WB et al: Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta 1: 3, 1980 |
|
Pijnenborg R, Robertson WB, Brosens I et al: Review article: Trophoblast invasion and the establishment of haemochorial placentation in man and laboratory animals. Placenta 2: 71, 1981 |
|
Brosens I, Robertson WB, Dixon HG: The physiological response to the vessels of the placental bed to normal pregnancy. J Pathol Bact 93: 570, 1967 |
|
Boyd JD, Hamilton WJ: Somite stages: The decidua capsularis. In The Human Placenta, Chap 7. Cambridge, England, W Heffer, 1970 |
|
Dhont M, De Sutter P, Ruyssinck G et al: Perinatal outcome of pregnancies after assisted reproduction: A case-control study. Am J Obstet Gynecol 181 (3): 688, 1999 |
|
Serour GI, Aboulghar M, Mansour R et al: Complications of medically assisted conception in 3,500 cycles. Fertil Steril 70 (4): 638, 1998 |
|
Hustin J, Schaaps JP: Echographic and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol 157: 162, 1987 |
|
Hustin J, Schaaps JP, Lambotte R: Anatomical studies of the uteroplacental vascularization in the first trimester of pregnancy. Troph Res 3: 49, 1988 |
|
Jauniaux E, Watson A, Burton G: Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks' gestation. Am J Obstet Gynecol 184 (5): 998, 2001 |
|
Jauniaux E, Watson AL, Hempstock J et al: Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol 157 (6): 2111, 2000 |
|
Jaffe R: Investigation of abnormal first-trimester gestations by color Doppler imaging. J Clin Ultrasound 21 (8): 521, 1993 |
|
Moll W: Invited Commentary: Absence of intervillous blood flow in the first trimester of human pregnancy. Placenta 16: 333, 1995 |
|
Craven CM, Zhao L, Ward K: Lateral placental growth occurs by trophoblast cell invasion of decidual veins. Placenta 21 (2–3): 160, 2000 |
|
Robertson WB, Brosens I, Dixon G: The Robertson-Brosens-Dixon hypothesis: Evidence for the role of haemochorial placentation in pregnancy success. Br J Obstet Gynaecol 98: 1195, 1991 |
|
Jurkovic D, Jauniaux E, Kurjak A et al: Transvaginal color Doppler assessment of the uteroplacental circulation in early pregnancy. Obstet Gynecol 77 (3): 365, 1991 |
|
Coppens M, Loquet P, Kollen M et al: Longitudinal evaluation of uteroplacental and umbilical blood flow changes in normal early pregnancy. Ultrasound Obstet Gynecol 7 (2): 114, 1996 |
|
Metcalfe J, McAnulty JH, Ueland K et al: Cardiovascular physiology. Clin Obstet Gynecol 24: 93, 1981 |
|
Khong TY, Sawyer IH, Heryet AR: An immunohistologic study of endothelialization of uteroplacental vessels in human pregnancy: Evidence that endothelium is focally disrupted by trophoblast in preeclampsia. Am J Obstet Gynecol 167: 751, 1992 |
|
Boyd JD, Hamilton WJ: Intervillous space. In The Human Placenta, Chap 18. Cambridge, England, W Heffer, 1970 |
|
Jauniaux E, Jurkovic D, Campbell S: Current Topic: In vivo investigation of the placental circulations by Doppler echography. Placenta 16: 323, 1995 |
|
Morizaki N, Morizaki J, Hayashi RH et al: A functional and structural study of the innervation of the human uterus. Am J Obstet Gynecol 160: 218, 1989 |
|
Reilly FD, Russell PT: Neurohistochemical evidence supporting an absence of adrenergic and cholinergic innervation in the human placenta and umbilical cord. Anat Rec 188: 277, 1977 |
|
Wilkin P: Pathologie du Placenta. Paris, Masson, 1965 |
|
Pijnenborg R, Bland JM, Robertson WB et al: Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta 4 (4): 397, 1983 |
|
Pijnenborg R, Bland JM, Robertson WB et al: The pattern of interstitial trophoblastic invasion of the myometrium in early human pregnancy. Placenta 2: 303, 1981 |
|
Kliman HJ, Feinberg RF: Human trophoblast-extracellular matrix (ECM) interactions in vitro: ECM thickness modulates morphology and proteolytic activity. Proc Natl Acad Sci U S A 87: 3057, 1990 |
|
Turpeenniemi-Hujanen T, Ronnberg L, Kauppila A et al: Laminin in the human embryo implantation: Analogy to the invasion by malignant cells. Fertil Steril 58: 105, 1992 |
|
Kovats S, Main EK, Librach C et al: A class I antigen, HLA-G, expressed in human trophoblasts. Science 248: 202, 1990 |
|
Kliman HJ, Feinberg RF: Differentiation of the trophoblast. Proc Natl Acad Sci U S A 87: 3, 1990 |
|
Enders AC: Trophoblast-uterine interactions in the first days of implantation: models for the study of implantation events in the human. Semin Reprod Med 18 (3): 255, 2000 |
|
Hemberger M, Cross JC: Genes governing placental development. Trends Endocrinol Metab 12 (4): 162, 2001 |
|
Kliman HJ: Uteroplacental blood flow. The story of decidualization, menstruation, and trophoblast invasion. Am J Pathol 157 (6): 1759, 2000 |
|
Boyd JD, Hamilton WJ: Structure and terminology of placental lobes and chorionic villi. In The Human Placenta, Chap 9. Cambridge, England, W Heffer, 1970 |
|
Vorherr H: Placental insufficiency and postmaturity. Eur J Obstet Gynecol Reprod Biol 5 (1–2): 109, 1975 |
|
Sands J, Dobbing J: Continuing growth and development of the third trimester human placenta. Placenta 6: 13, 1985 |
|
Salafia CM, Weigl CA, Foye GJ: Correlation of placental erythrocyte morphology and gestational age. Pediatr Pathol 8: 495, 1988 |
|
Goldstein J, Braverman M, Salafia C et al: The phenotype of human placental macrophages and its variation with gestational age. Am J Pathol 133: 648, 1988 |
|
Bernirschke K, Kaufmann P (eds): Pathology of the Human Placenta. New York, Springer-Verlag, 1990 |
|
Boyd JD, Hamilton WJ: General descriptions of specimens. In The Human Placenta, Chap 8. Cambridge, W Heffer, 1970 |
|
Schneider H: Current Topic: Ontogenic changes in the nutritive function of the placenta. Placenta 17: 15, 1996 |
|
Molteni RA: Placental growth and fetal/placental weight (F/P) ratios throughout gestation: Their relationship to patterns of fetal growth. Semin Perinatol 8 (2): 94, 1984 |
|
Teasdale F, Jean-Jacques G: Morphometric evaluation of the microvillous surface enlargement factor in the human placenta from mid-gestation to term. Placenta 6: 375, 1985 |
|
Fenley MR, Burton GI: Villous composition and membrane thickness in the human placenta at term: A stereological study. Placenta 12: 131, 1991 |
|
Teasdale F: Gestational changes in the functional structure of the human placenta in relation to fetal growth: A morphometric study. Am J Obstet Gynecol 137: 560, 1980 |
|
Siddiqi TA, Meyer RA, Lych-Salamon D et al: A prospective longitudinal study of human umbilical arterial blood flow (abstr 464). Am J Obstet Gynecol 167 (suppl): 170, 1994 |
|
Bonds DR, Crosby LO, Cheek TG et al: Estimation of human fetal-placental unit metabolic rate by application of the Fick principle. J Dev Physiol 8: 45, 1986 |
|
Towstoless MK, Congiu M, Coghlan JP et al: Placental and renal control of plasma osmolality in chronically cannulated ovine fetus. Am J Physiol 253: R389, 1987 |
|
Bell RJ, Congiu M, Hardy KJ et al: Gestation dependent aspects of the response of the ovine fetus to the osmotic stress induced by maternal water deprivation. Q J Exp Physiol 69: 187, 1984 |
|
Wintour EM: Water metabolism in the fetal placental unit. In Cowett RM (ed): Principles of Perinatal-Neonatal Metabolism, Chap 19. New York, Springer-Verlag, 1991 |
|
Longo LD: Respiration in the fetal placental unit. In Cowett RM (ed): Principles of Perinatal-Neonatal Metabolism, Chap 7. New York, Springer-Verlag, 1991 |
|
Clapp JF, Szeto HH, Larrow R et al: Fetal metabolic response to experimental placental vascular damage. Am J Obstet Gynecol 140: 446, 1981 |
|
Kurman RJ: The morphology, biology and pathology of intermediate trophoblast: A look back to the present. Hum Pathol 22: 847, 1991 |
|
Vassilakos P, Kajii T: Hydatidiform mole: Two entities. Lancet 1: 259, 1976 |
|
Vassilakos P, Riotton G, Kajii T: Hydatidiform mole: Two entities, a morphologic and cytogenetic study with some clinical considerations. Am J Obstet Gynecol 127: 167, 1977 |
|
Szulman AE, Surti U: The syndromes of hydatidiform mole: I. Cytogenetic and morphologic correlation. Am J Obstet Gynecol 131: 655, 1987 |
|
Szulman AE, Surti U: The syndromes of hydatidiform mole: II. Morphologic evolution of the complete and partial mole. Am J Obstet Gynecol 132: 20, 1978 |
|
Lage JM: Diagnostic dilemmas in gynecologic and obstetric pathology. Semin Diagn Pathol 7: 146, 1990 |
|
Szulman AE: Trophoblastic disease: Clinical pathology of hydatidiform moles. Obstet Gynecol Clin North Am 15: 443, 1988 |
|
Conran RM, Hitchcock CL, Popek EJ et al: Diagnostic considerations in molar gestations. Hum Pathol 24: 41, 1993 |
|
Kajii T, Ohama K: Androgenetic origin of hydatidiform mole. Nature 268: 633, 1977 |
|
Robert DJ, Mutter GL: Advances in the molecular biology of gestational trophoblastic disease. J Reprod Med 139 (3): 201, 1994 |
|
Genest DR, Oaborde O, Berkowitz RS et al: A clinicopathologic study of 153 cases of complete hydatidiform mole (1980-1990): Histologic grade lacks prognostic significance. Obstet Gynecol 78: 402, 1991 |
|
Keep D, Varagoza MV, Hassold T et al: Very early complete hydatidiform mole. Hum Pathol 27: 708, 1996 |
|
Wake N, Jujino T, Hoshi S et al: The propensity to malignancy of dispermic heterozygous moles. Placenta 8: 319, 1987 |
|
Surti V: Genetic concepts and techniques. In Sulzman AE, Buchsbaum HJ (ed): Gestational Trophoblastic Disease. New York, Springer-Verlag, 1987 |
|
Szulman AE, Philippe E, Boue JG et al: Human triploidy: Association with partial hydatidiform moles and nonmolar conceptuses. Hum Pathol 12: 1016, 1981 |
|
Davis JR, Kerrigan DP, Way DL et al: Partial hydatidiform moles: Deoxyribonucleic acid content and course. Am J Obstet Gynecol 157: 969, 1987 |
|
Teng NNH, Ballon SC: Partial hydatidiform mole with diploid karyotype: Report of three cases. Am J Obstet Gynecol 150: 961, 1984 |
|
Gardner HAR, Lage JM: Choriocarcinoma following a partial hydatidiform mole: A case report. Hum Pathol 23: 468, 1992 |
|
Heifitz SA, Czaja J: In situ choriocarcinoma arising in partial hydatidiform mole: Implications for the risk of persistent trophoblastic disease. Pediatr Pathol 12: 601, 1992 |
|
Bagshawe KD, Lawler SD, Paradinas FJ et al: Gestational trophoblastic tumors following initial diagnosis of partial hydatidiform mole. Lancet 335: 1074, 1990 |
|
Salafia CM, Maier D, Vogel C et al: Placental and decidual histology in spontaneous abortions: Detailed description and correlations with chromosome number. Obstet Gynecol 82: 295, 1993 |
|
Van Lijnschoten G, Arends JW, Thunnissen FBJM et al: A morphometric approach to the relation of karyotype, gestational age, and histological features in early spontaneous abortions. Placenta 15: 189, 1994 |
|
Rockelein G, Ulmer R, Schroder J: Karyotype and placental structure of first trimester spontaneous abortions: A morphometrical study. Eur J Obstet Gynecol Reprod Biol 38: 25, 1990 |
|
Genest DR, Roberts D, Boyd T et al: Fetoplacental histology as a predictor of karyotype: A controlled study of spontaneous first trimester abortions. Hum Pathol 26: 201, 1995 |
|
Redline RW, Zaragoza M, Hassold T: Prevalence of developmental and inflammatory lesions in nonmolar first-trimester spontaneous abortions. Hum Pathol 30 (1): 93, 1999 |
|
Hassold TJ: A cytogenetic study of repeated spontaneous abortions. Am J Hum Genet 32: 723, 1980 |
|
Warburton D, Kline J, Stein Z et al: Does the karyotype of a spontaneous abortion predict the karyotype of a subsequent abortion? Evidence from 273 women with two karyotyped spontaneous abortions. Am J Hum Genet 41: 465, 1987 |
|
Hecht F, Hecht BK, Berger CS: Aneuploidy in recurrent spontaneous aborters: The tendency to parental non-disjunction. Clin Genet 26: 43, 1984 |
|
Abramowsky CR, Vegas ME, Swinehart G et al: Decidual vasculopathy of the placenta in lupus erythematosus. N Engl J Med 303: 668, 1980 |
|
Russell P, Atkinson K, Krishnan L: Recurrent reproductive failure due to severe placental villitis of unknown etiology. J Reprod Med 24 (2): 93, 1980 |
|
Doss BJ, Green MF, Hill J et al: Massive chronic intervillositis associated with recurrent abortions. Hum Pathol 26: 1245, 1995 |
|
Cowchock S, Dehoratius RD, Wapner RJ et al: Subclinical autoimmune disease and unexplained abortion. Am J Obstet Gynecol 150: 367, 1984 |
|
Scott JR, Rote NS, Branch DW: Immunologic aspects of recurrent abortion and fetal death. Obstet Gynecol 70: 645, 1987 |
|
Salafia CM, Parke AL, Starzyk KA et al: Obstetric complications in the APL syndrome. In Ascherson R (ed): The APL Syndrome. Boca Raton, CRC Press, 1996 |
|
Grimmer D, Landas S, Kemp JD: IgM antitrophoblast antibodies in a patient with a pregnancy-associated lupuslike disorder, vasculitis, and recurrent intrauterine fetal demise. Arch Pathol Lab Med 112: 191, 1988 |
|
Kalousek DK: The role of confined chromosomal mosaicism in placental function and human development. Growth Gen Horm 4 (4): 1, 1988 |
|
Honore LH, Dill FJ, Poland BJ: Placental morphology in spontaneous human abortuses with normal and abnormal karyotypes. Teratology 14: 151, 1976 |
|
Rochelson B, Kaplan C, Guzman E et al: A quantitative analysis of placental vasculature in the third trimester fetus with autosomal trisomy. Obstet Gynecol 75: 59, 1990 |
|
Krause WW: [Diphtheria as a general infection.] Med Klin. 1950 Apr 7;45(14):417-22. |